Analysis Tutorial: Interpreting Rosetta Outputs
KEYWORDS: ANALYSIS GENERAL
This tutorial will go through some examples of what to do with the output that Rosetta produces. In general the output is one or several of the following files:
- pdb file - protein structural information in the Protein DataBank format
- silent file - protein structural information in Rosetta's own format
- score file - tab-delimited sets of scores
- log file - a record of all the output to terminal by a given run
IMPORTANT Rosetta will always give you a result!
The main question that you have to answer is:
"Does the result answer my question?"
This tutorial is primarily focused on statistical and analytical methods to inform the answer to that question. As with any stochastic simulation, many of the results Rosetta generates for any given run will explore regions of conformation space that are not relevant to the problem at hand; statistical analysis can help select the best/most informative results from a given run while also evaluating the success of the run as a whole.
PDB file
The models that Rosetta produces are typically in PDB file format. If you open the file, you will of course find all atom positions. Additionally, Rosetta will append all scoring terms for every residue at the end of the pdb file.
-
Score the provided pdb file and open the scored structure in your favourite text editor.
$> ../../../main/source/bin/score_jd2.default.linuxclangrelease -s 1ubq.pdb -out:output -out:file:scorefile score.sc -
Now, search for the word "pose". This will bring you to the end of the coordinate section. It should look similar to this:
# All scores below are weighted scores, not raw scores. #BEGIN_POSE_ENERGIES_TABLE S_0004_design_0062.pdb label fa_atr fa_rep fa_sol fa_intra_rep fa_elec pro_close hbond_sr_bb hbond_lr_bb hbond_bb_sc hbond_sc dslf_fa13 rama omega fa_dun p_aa_pp ref total weights 0.8 0.44 0.75 0.004 0.7 1 1.17 1.17 1.17 1.1 1 0.2 0.5 0.56 0.32 1 NA pose -513.648 51.38 327.036 1.19669 -61.0293 2.86533 -26.313 -42.5744 -9.85907 -17.8907 -11.8656 -9.94805 10.566 150.226 -17.7945 -29.4897 -197.143 THR:NtermProteinFull_1 -2.22882 0.1511 2.60394 0.00934 -0.65245 0 0 0 -0.29914 -0.28075 0 0 0.10172 0.02367 0 0.16454 -0.40685 VAL_2 -2.91409 0.33743 1.30547 0.01945 -0.14243 0 0 0 0 0 0 -0.10417 0.10036 0.61358 0.17219 0.74484 0.13264line starts with What does this mean? label names of the included energy terms (scores) weights every scoring term has a weighting factor pose every scoring term as the sum of all residues THR:NtermP[...] scores for residue #1 (N-terminus, threonine) VAL_2 scores for residue #2 (here a Valine) -
Extract the total score for each individual residue using egrep:
$ egrep '[^A-Z]*_[0-9]+\s' 1ubq_0001.pdb | cut -d'_' -f2 | awk '{print $1 "\t" $NF}' > total_score_per_res.datThis selects all lines that contains the pattern "Letters_[some-number] ", then gets rid of everything before the underscore and finally prints out the first field (the number after _), a tab, and the very last field (here: the total_score)
Now, you can plot the residue position vs. total score, e.g. as a histogram. You should notice that there are regions with relatively bad energies (around positions 35-40).
The score file
The score file contains the scores for all models generated in a run, broken down into the individual terms. You can extract certain columns (i.e. scoring terms) or sort them by any column.
$ sort -n -k2 example_score_file.sc
this will sort the entire file by column 2 (total score)
$ sort -n -k2 example_score_file | awk '{print $2 "\t" $3}'
this will do the same, but only print out columns 2 and 3
-
Extract score and rmsd values for the best 1000 models!
$ sort -n -k2 example_score_file.sc | head -n 1000 | awk '{print $2 "\t" $25 "\t" $NF}' > score_rmsd.dat
this will sort by total score, take only the top 1000, extract columns 2 (score), 25 (rms) and the very last one (description,tag) and write this into a new file, called score_rmsd.dat.
Note: The position of scoring terms in the score file depends on what protocol you are running and what options you are using!
- Plot column 2 (rmsd) vs column 1 (score) of score_rmsd.dat
Having gnuplot installed makes it easy to make quick plots. But you can use any software to plot the two columns.
$ gnuplot
Terminal type set to 'x11'
gnuplot> plot "score_rmsd.dat" u 2:1
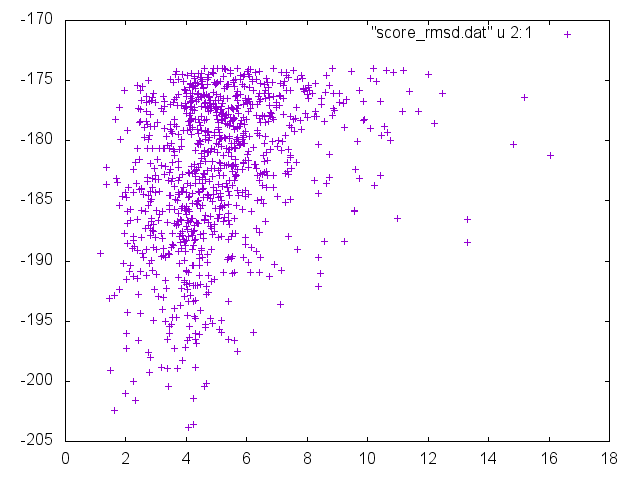
You can see that the lower energy values (y-axis) correspond to lower rmsds (x-values). That means that there is a significant degree of convergence.
Extracting silent files
When you produce a large number of models (e.g. for de novo structure prediction), it is recommended to output the models as binary silent files:
-out:file:silentfile_struct_type binary
-out:file:silent <file name of your choice>
Rosetta provides multiple ways to extract structures from those files.
1. Scoring - The scoring application can use silent files as input and generates pdb files.
Try the following:
$> ../../../main/source/bin/score_jd2.default.linuxgccrelease -in:file:silent_struct_type binary -in:file:silent example.out -out:pdb -out:file:scorefile extracted_scorefile.sc @general_flags
this will give you a new scorefile and a PDB for each structure in the silent file (here there is only one structure).
For scoring particular structures, you can add:
-in:file:tags S_00000170_1
This can take multiple tags (e.g. -in:file:tags S_00000170_1 S_00000170_2 S_00000168_1)
2. Extract PDBs application
If you ran the previous example, first remove the extracted pdb file:
$ rm S_00000170_1.pdb
Then try this:
$> ../../../main/source/bin/extract_pdbs.linuxgccrelease -in:file:silent_struct_type binary -in:file:silent example.out
You should now have the pdb extracted. The -in:file:tags option works here, too.
You can get the best energy models by sorting the score file, identifying the top structures, and then extracting them using their tags.
Clustering
For a result to be reasonably reliable, you should always get multiple similar solutions with similar energies. Outliers do appear - structures with artificially good scores. To find out whether your low energy solutions are outliers or have been sampled multiple times with slight very differences in structure and energy, clustering the models by structural similarity is extremely useful.
While Rosetta has a clustering applications, its use is very limited. Other, more sophisticated clustering software exists that should be used in preference.
More information on clustering can also be found in the Rosetta manual.
Controls
A good way to evaluate whether you can trust your results from Rosetta is to include control runs. Often you might find homologous proteins, protein complexes, peptides, etc. similar to your unknown system, but where the experimental results of interest are know. Using those as positive/negative controls is a good idea. For example, if the peptide TRRTFGAH is known to bind, but Rosetta fails to dock it correctly, then you might not want to trust your docking results for TRRSYGAH. Conversely, if the peptide TLLTFGAH is known not to bind, but Rosetta cannot distinguish between the binding of TLLTFGAH and TRRTFGAH, then the protocol may not have the accuracy or dynamic range to rank the binding of peptides of unknown affinities.
Compare with experimental results
If you're attempting to predict a quantitative result with a Rosetta protocol, you should first plot a calibration curve based on similar systems with known results. Rosetta's energy results are in arbitrary units, and the results will be sensitive to the fine details of a particular protocol. A correlation may exist (for example, there is a correlation between (de)stabilizing and Rosetta's predicted ddG), but the details of the correlation will be sensitive to exact protocol details. Running the calculation on systems with known experimental results, and seeing the correlation between predicted and experimental value will allow you to 1. estimate the experimental value from the predicted results, and 2. give you an estimate on the prediction accuracy of the protocol. A very noisy correlation means that the accuracy of the prediction is questionable - you should be skeptical of the predictions which Rosetta produces.