Full Atom Representation vs Centroid Representation
KEYWORDS: CORE_CONCEPTS ANALYSIS
Tutorial by Shourya S. Roy Burman (ssrb@jhu.edu) Created 20 June 2016
The demos are available at <path_to_Rosetta_directory>/demos/tutorials/full_atom_vs_centroid. All demo commands listed in this tutorial should be executed when in this directory. All the demos here use the linuxgccrelease binary. You may be required to change it to whatever is appropriate given your operating system and compiler.
Summary
Rosetta uses two primary representations while working with protein structures — full atom and centroid. By the end of this tutorial, you should understand:
- The need for the centroid representation
- How centroid representation differs from full atom
- How to convert your structure from one representation to the other
- How to identify when a protocol requires a particular representation and you have provided the other one
Need for a reduced representation
In an ideal world with infinite time and computer power, we could perform all of our simulations with all atoms. In practice, trying to perform extensive backbone sampling while also including all sidechain atoms is impractical at best.
The first problem is that having all atoms is expensive, because calculating interactions between all atom pairs grows rapidly (n2) with the number of atoms. The bigger problem is that fully atomic conformational space is very rugged, so most moves are rejected by Monte Carlo.
Centroid representation
To get around this problem, poses are often converted into centroid mode for portions of a protocol that require extensive backbone sampling (for example, the initial stages of de novo structure prediction. In centroid mode, the backbone remains fully atomic, but the representation of each sidechain is simplified to a single pseudo-atom of varying size. For protein backbones, this representation preserves five backbone atoms for each amino acid: nitrogen (N), the alpha carbon (CA), the carbonyl carbon (C), the carbonyl oxygen (O), and the polar hydrogen on nitrogen. The sidechain is replaced by the beta carbon (CB) and a CEN atom whose radius and properties (polarity, charge, etc.) are determined by the residue's identity.
Centroid score function
Centroid score functions typically have fewer and simpler energy terms than full atom score functions. The following are a list of terms in the optimized cen_std_smooth score function:
cen_env_smooth context-dependent one-body energy term that describes the solvation of a particular residue (based on the hydrophobic effect)
cen_pair_smooth two-body energy term for residue pair interactions (electrostatics and disulfide bonds)
cbeta_smooth solvation term intended to correct for the excluded volume effect introduced by the simulation and favor compact structures
vdw represents only steric repulsion and not attractive van der Waals' forcesOwing to the simplified scoring, this has a disadvantage in terms of interpreting results, but a huge advantage in that the energy landscape is not nearly as rugged, and sampling very different conformations is easier.
Returning to the full atom representation
After large-scale sampling in centroid mode, poses are generally converted back to their all-atom representation for refinement. The centroid to full atom conversion involves placing the atoms which are missing in the centroid representation. Simply placing the atoms in a "default" conformation without regards to the residue environment is likely to result in unsatisfactory structures. Therefore, the centroid to full atom conversion generally entails some combination of sidechain repacking and minimization to optimize the locations of the added atoms. This allows Rosetta to more accurately score interactions between sidechains and other finer details of the protein's structure.
Example
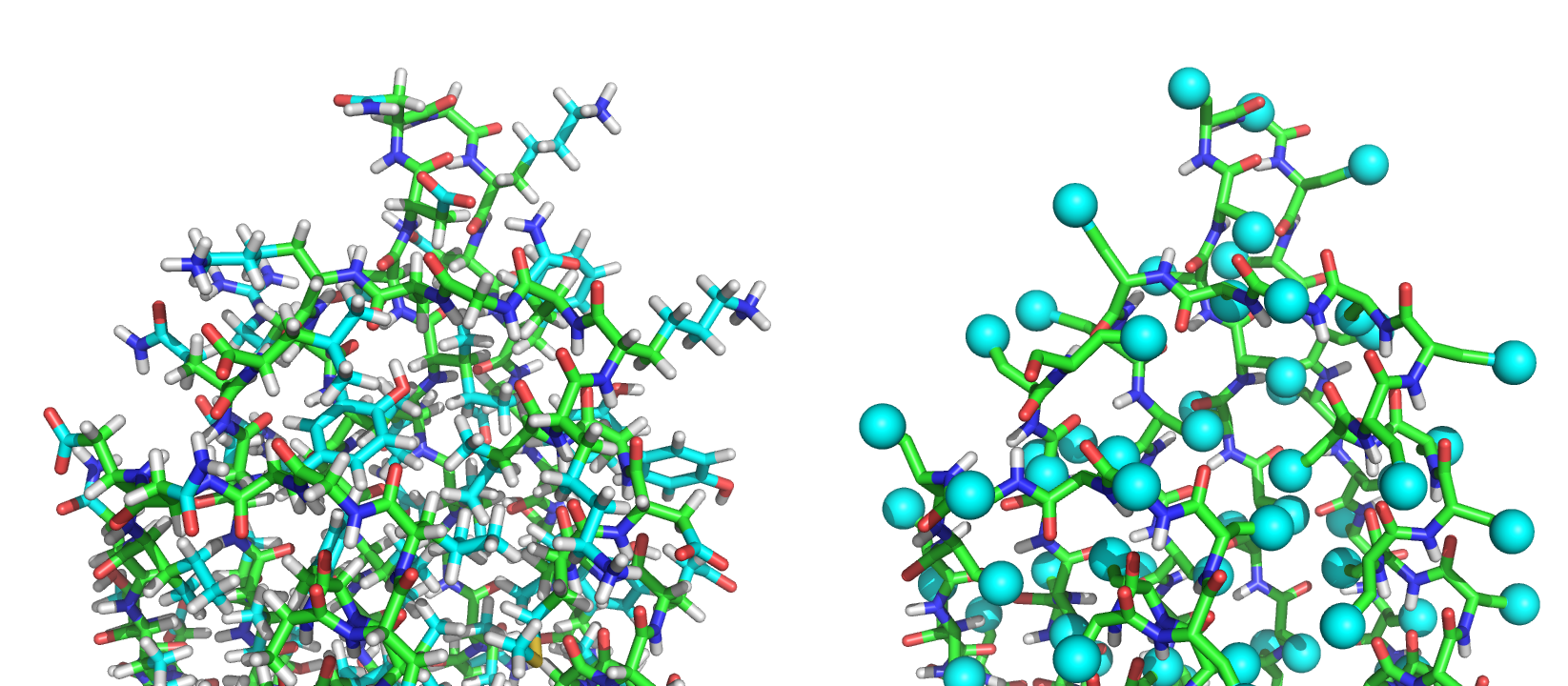 The PDB 1QYS in the (L) full atom representation, and (R) the centroid representation
The PDB 1QYS in the (L) full atom representation, and (R) the centroid representation
PDB files generated in the centroid format have the sidechain atoms (Cγ onwards) replaced by one CEN pseudo-atom. For example, the first two residues of the PDB 1QYS in full atom look like:
ATOM 1 N ASP A 3 -4.524 18.589 17.199 1.00 0.00 N
ATOM 2 CA ASP A 3 -3.055 18.336 17.160 1.00 0.00 C
ATOM 3 C ASP A 3 -2.676 17.087 16.375 1.00 0.00 C
ATOM 4 O ASP A 3 -3.539 16.391 15.835 1.00 0.00 O
ATOM 5 CB ASP A 3 -2.498 18.208 18.580 1.00 0.00 C
ATOM 6 CG ASP A 3 -3.070 17.016 19.336 1.00 0.00 C
ATOM 7 OD1 ASP A 3 -3.497 16.083 18.699 1.00 0.00 O
ATOM 8 OD2 ASP A 3 -3.073 17.050 20.543 1.00 0.00 O
ATOM 9 1H ASP A 3 -4.705 19.419 17.727 1.00 0.00 H
ATOM 10 2H ASP A 3 -4.868 18.706 16.268 1.00 0.00 H
ATOM 11 3H ASP A 3 -4.985 17.814 17.630 1.00 0.00 H
ATOM 12 HA ASP A 3 -2.571 19.180 16.669 1.00 0.00 H
ATOM 13 1HB ASP A 3 -1.413 18.107 18.538 1.00 0.00 H
ATOM 14 2HB ASP A 3 -2.720 19.116 19.141 1.00 0.00 H
...The first two residues of 1QYS in the centroid mode look like:
ATOM 1 N ASP A 3 -4.524 18.589 17.199 1.00 0.00 N
ATOM 2 CA ASP A 3 -3.055 18.336 17.160 1.00 0.00 C
ATOM 3 C ASP A 3 -2.676 17.087 16.375 1.00 0.00 C
ATOM 4 O ASP A 3 -3.539 16.391 15.835 1.00 0.00 O
ATOM 5 CB ASP A 3 -2.496 18.220 18.580 1.00 0.00 C
ATOM 6 CEN ASP A 3 -2.022 18.783 19.285 1.00 0.00 X
ATOM 7 H ASP A 3 -5.003 18.619 18.076 1.00 0.00 H
...The full PDBs are available at <path_to_Rosetta_directory>/demos/tutorials/full_atom_vs_centroid/input_files.
Using the wrong representation
Centroid input when protocol expects full atom
The following demo runs the scoring protocol (which expects a full atom input) with an centroid PDB as input.
$> ../../../main/source/bin/score_jd2.linuxgccrelease @flag_cen_for_fa
If we provide a centroid PDB input to a protocol that expects a full atom input, typically, the program does not crash. Instead, Rosetta first discards the centroid pseduo-atoms and displays the following warnings:
...
core.io.pose_from_sfr.PoseFromSFRBuilder: [ WARNING ] discarding 1 atoms at position 1 in file input_files/1qys_centroid.pdb. Best match rsd_type: ASP:NtermProteinFull
core.io.pose_from_sfr.PoseFromSFRBuilder: [ WARNING ] discarding 1 atoms at position 2 in file input_files/1qys_centroid.pdb. Best match rsd_type: ILE
...Then, Rosetta realizes that the sidechains are missing.
...
core.conformation.Conformation: [ WARNING ] missing heavyatom: CG on residue ASP:NtermProteinFull 1
core.conformation.Conformation: [ WARNING ] missing heavyatom: OD1 on residue ASP:NtermProteinFull 1
core.conformation.Conformation: [ WARNING ] missing heavyatom: OD2 on residue ASP:NtermProteinFull 1
core.conformation.Conformation: [ WARNING ] missing heavyatom: CG1 on residue ILE 2
core.conformation.Conformation: [ WARNING ] missing heavyatom: CG2 on residue ILE 2
core.conformation.Conformation: [ WARNING ] missing heavyatom: CD1 on residue ILE 2
...Finally, Rosetta builds the missing sidechains.
...
core.pack.pack_missing_sidechains: packing residue number 1 because of missing atom number 6 atom name CG
core.pack.pack_missing_sidechains: packing residue number 2 because of missing atom number 6 atom name CG
...Since there is no information about the conformation of the sidechains in the centroid representation, different runs produce slightly different sidechain conformations.
Full atom input when protocol expects centroid
The following demo runs the scoring protocol with an option to score the structure assuming it to be in centroid, but the input PDB supplied is full atom:
-in:file:centroid
-score:weights cen_std_smooth Run:
$> $ROSETTA3/bin/score_jd2.linuxgccrelease @flag_fa_for_cenIf we provide a full atom PDB input to a protocol that expects a centroid input, typically the program does not stop. Instead, Rosetta first discards all sidechain atoms beyond Cβ and displays the following warnings:
...
core.io.pose_from_sfr.PoseFromSFRBuilder: [ WARNING ] discarding 9 atoms at position 1 in file input_files/1qys.pdb. Best match rsd_type: ASP:NtermProteinFull
core.io.pose_from_sfr.PoseFromSFRBuilder: [ WARNING ] discarding 13 atoms at position 2 in file input_files/1qys.pdb. Best match rsd_type: ILE
...Then, Rosetta realizes that the centroid pseudo-atoms are missing.
...
core.conformation.Conformation: [ WARNING ] missing heavyatom: CEN on residue ASP:NtermProteinFull 1
core.conformation.Conformation: [ WARNING ] missing heavyatom: CEN on residue ILE 2
...Converting from one representation to the other
There is not a dedicated executable to switch one representation to the other. Instead, the conversion is normally a function of Rosetta's structure input mechanism. In this section, we will exploit the automatic residue building functionality of the scoring protocol to convert from centroid to full atom. To convert full atom to centroid, we will write a short XML script using RosettaScripts.
Converting from full atom to centroid and back will not give you back the same structure, as sidechain building in Rosetta is not deterministic. However, centroid->fullatom->centroid should return the original structure.
There are at least two ways to convert the representation. One is shown above: by using scoring application. To obtain a pdb file, you need to add the output option -out:output
A second option that is good to know about is using RosettaScripts. This way, converting representations can be part of a larger Rosetta Script. The following section will demonstrate how to do that.
Converting from centroid to full atom
To convert a full atom PDB to centroid, we need to interface with Rosetta at a level deeper than any executable will allow us to do. The simplest way to do this is to write a script using RosettaScripts. The following short XML script calls a mover (SwitchResidueTypeSetMover) and asks it to convert the centroid structure to full atom:
<ROSETTASCRIPTS>
<SCOREFXNS>
</SCOREFXNS>
<FILTERS>
</FILTERS>
<MOVERS>
<SwitchResidueTypeSetMover name="switch_repr" set="fa_standard" />
</MOVERS>
<APPLY_TO_POSE>
</APPLY_TO_POSE>
<PROTOCOLS>
<Add mover="switch_repr" />
</PROTOCOLS>
</ROSETTASCRIPTS>The Rosetta Scripts application is then invoked like:
$> $ROSETTA3/bin/rosetta_scripts.linuxgccrelease @flag_from_cen_to_faNOTE: The file in
output_files/has the word centroid in it because the input file did, not because it is in centroid mode!
Compare this to the PDB <path_to_Rosetta_directory>/demos/tutorials/full_atom_vs_centroid/output_files/expected_output/1qys_centroid_0001.pdb. You will notice that the files have different sidechain orientations, but the same backbone atom positions.
Converting from full atom to centroid
Almost the same script can do the reverse operation:
<ROSETTASCRIPTS>
<SCOREFXNS>
</SCOREFXNS>
<FILTERS>
</FILTERS>
<MOVERS>
<SwitchResidueTypeSetMover name="switch_repr" set="centroid" />
</MOVERS>
<APPLY_TO_POSE>
</APPLY_TO_POSE>
<PROTOCOLS>
<Add mover="switch_repr" />
</PROTOCOLS>
</ROSETTASCRIPTS>This can be found at <path_to_Rosetta_directory>/demos/tutorials/full_atom_vs_centroid/fa_to_cen.xml.
Run in the terminal:
$> ../../../main/source/bin/rosetta_scripts.linuxgccrelease @flag_from_fa_to_cen
This should produce a centroid file output_files/1qys_0001.pdb
Compare this to the PDB output_files/expected_output/1qys_0001.pdb. The files should be exactly the same.