Getting started
This page is written for an audience of scientists new to Rosetta: perhaps a first year graduate student, or young postdoc, who has received/started a project that needs "some computer modeling". In other words, an individual coming to Rosetta from a cold start. Is Rosetta a good tool for the modeling you need to do? If so, how do you go about getting and using Rosetta? If you are already comfortable with the concepts, feel free to skip ahead.
Rosetta is a very large software suite for macromolecular modeling. By software suite, we mean that it is a large collection of computer code (mostly in C++, some in Python, a little in other languages), but it is not a single monolithic program. By macromolecular modeling, we mean the process of evaluating and ranking the physical plausibility of different structures of biological macromolecules (usually protein, but nucleic acids and ligands are significantly supported and support for implicit lipid membranes is growing). Generally, a user will choose some specific protocol within Rosetta and provide that protocol with inputs for A) what structure to work on, and B) what options within the protocol are appropriate for the user's needs.
Do I have what I need?
Doing macromolecular modeling well—doing good science—requires careful consideration of your inputs, how the modeling is performed, and analysis of your outputs. Rosetta itself can be operated as a "black box", but you are doing yourself and your science a disservice if you use it that way.
1) Inputs to Rosetta
The major input to Rosetta is the input structure. Generally, if you have a high-resolution structure(s) (better than 2 Å) of your molecule, it can be used with Rosetta with few changes. If you have a poorer-resolution structure, an NMR structure, a homology model, or no structure at all, then your task will be much harder. You should still prepare your structure for modeling, but be aware that modeling is less efficient and effective when starting from poor quality structures and interpreting results is more challenging.
2) Choosing the Rosetta protocol
The other inputs are the choice of which Rosetta protocol to use, and what options or file inputs to use.
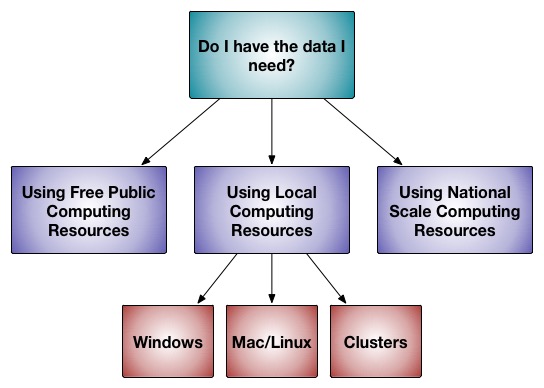
3) Computational resources
Rosetta software, as a whole, is written to run on supercomputers, but can be run on many different scales. Most applications can give "dry runs" for testing on any computer. A few applications can run on laptop computers. There are applications than can run on lab-scale powerful computers (12-30 core range). Most applications assume you have access to tens of thousands of hours of computer time to accumulate enough results to answer your question. The later sections of this document describe installing or using Rosetta at those different scales.
Public Rosetta Servers
RosettaCommons maintains several public servers for NON-COMMERCIAL USE ONLY. The servers of broadest interest are touched on here, but see here and here for more complete lists.
For commercial use, see the section below and the Rosetta Servers page.
ROSIE is a server that offers several Rosetta applications through a simple web interface. It is perfect for use by those new to Rosetta. Despite ROSIE's variety it offers only a slice of Rosetta's full functionality.
ROBETTA (Robot-Rosetta) is a server that provides ab initio folding and structure prediction, as well as fragment selection for local runs of Rosetta.
Commercial Rosetta Servers
- Cyrus Biotechnology is a company that offers Bench, a server for commercial users that has tools for homology modeling (like Robetta), protein design (RosettaDesign), ddG calculation, and other modeling tools like relaxation and minimization. (Cyrus Biotechnology is a commercial Rosetta licensee offering a web-based graphical user interface for Rosetta to its customers. Licensing fees paid by commercial Rosetta licensees to the University of Washington are used to support the RosettaCommons, investing in the maintenance and further development of Rosetta.)
Local installation and use of Rosetta
Rosetta cam be downloaded from https://rosettacommons.org/software/download/. Academic and non-commercial users are automatically granted a non-commercial-use-only license with the download. Commercial use requires a paid license obtainable from UW CoMotion.
Note that the download comes as a tar archive. Pre-compiled executables are available for some platforms. Source code is also availible, such that you can compile on platforms where pre-compiled executables are not availible.
Introductory Rosetta Tutorials
After downloading Rosetta, go through these detailed tutorials to get started. Once you are done with the introductory tutorials, you should be able to do most common molecular modeling tasks on Rosetta.
Installation on Mac/Linux
Local installation implies that one will be using Rosetta through a command line interface (or terminal). For local users, you are unlikely to want to install Rosetta to the entire system. Rosetta is quite happy to be compiled and installed by regular users without administrative rights — this is how the developers use it. You may need administrative rights to install dependencies.
First untar/uncompress your downloaded copy of the code (
tar -xvzf Rosetta[releasenumber].tar.gz).Next, navigate to the
sourcedirectory:cd main/source.Rosetta uses SCons as a compile assistant. Rosetta comes with a copy of SCons, which should work for most systems.
The basic compilation command is
./scons.py -j<number_of_processors_to_use> mode=release bin. Replace with a number one processor fewer than your computer has. Expect compilation to take a while (hours on one processor).
See our extensive build documentation for further instructions and troubleshooting.
Windows
Unfortunately, we are not currently able to support the whole of Rosetta on Windows. There are few free, easy-to-use C++ compilers available for Windows, and they use slightly different C++ standards. Dual booting or virtual machines running Linux/MacOS are options. We cannot help set up Windows/Linux dual boots, but we can help with Rosetta on the Linux partition. A subset of Rosetta that is required by PyRosetta, a Python-based interface to Rosetta, is supported on Windows and made available on http://www.pyrosetta.org.
Use on supercomputer clusters
If you'll be running Rosetta on a scientific computation cluster, there may already be a version of Rosetta installed for general usage. Talk to your cluster administrator to see if there is a centrally-provided version available for your use.
If your cluster doesn't have Rosetta already installed, or you wish to use a different version than the centrally installed one, don't worry — Rosetta is designed to be compiled and installed by regular users without administrative rights. As long as commonly available compilation tools are available for your use, you should be able to build and run Rosetta in your user directory without cluster administrator involvement. Just treat it like a local install into userspace.
Use of Rosetta on national-scale supercomputing resources
As part of the XSEDE initiative, the TACC/Stampede cluster has Rosetta and PyRosetta centrally installed for authorized users. See the TACC page for more details.
How to update these docs
Developers can update these doc pages through our interactive wiki [note: Github login required], or anyone can suggest changes through the Github repository.
See Also
- Introductory Rosetta Tutorials
- Resources for learning biophysics and computational modeling
- Build Documentation: Information on setting up Rosetta
- I want to do x: Guides to specific types of structural perturbations using RosettaScripts
- Application Documentation: Links to documentation for a variety of Rosetta applications
- Analyzing Results: Tips for analyzing results generated using Rosetta
- Comparing structures: Essay on comparing structures
- Commands collection: A list of example command lines for running Rosetta executable files
- Rosetta Servers: Web-based servers for Rosetta applications
- Scripting Documentation: Scripting interfaces to Rosetta
- Rosetta overview: Overview of major concepts in Rosetta
- FAQ: Frequently asked questions
- Glossary: Brief definitions of Rosetta terms
- RosettaEncyclopedia: Detailed descriptions of Rosetta terms