Fragment-free helical bundle structure prediction using the helical_bundle_predict application
Back to Application Documentation.
Created 21 August 2019 by Vikram K. Mulligan, Flatiron Institute (vmulligan@flatironinstitute.org). Last updated 14 November 2020.
This application is currently unpublished! If you use this application, please include the developer in the list of authors for your paper.
Description
Rosetta's most widely-used structure prediction application, Rosetta ab initio, relies on fragments of proteins of known structure to guide the search of the conformation space, and to limit the conformational search to the very small subset of the space that resembles known protein structures. This works reasonably well for natural proteins, but prevents the prediction of structures built from non-natural building blocks. In 2015, we introduced the simple_cycpep_predict application to allow the prediction of structures of small heteropolymers built from any mixture of chemical building-blocks, without any known template sequences. The simple_cycpep_predict application uses the generalized kinematic closure algorithm to confine the search to closed conformations of a macrocycle, effectively limiting the search space without relying on databases of known structures. Unfortunately, this only works for relatively small molecules (less than approx. 15 residues), molecules with regions of known secondary structure (less than approx. 10 residues of loop), or molecules with internal symmetry (less than approx. 8 residues in the repeating unit), and absolutely requires covalent linkage between the ends of the molecule. A prediction strategy for larger, linear heteropolymers built from non-natural building-blocks is needed.
The helical_bundle_predict application was created to fill this niche. Based on the hypothesis that fragments are primarily useful for sampling allowed bends of known secondary structures (which can be sampled using parametric approaches, given heteropolymer secondary structures that can be predicted a priori) or allowed loop conformations (which can be sampled using random perturbations or kinematic approaches), this application carries out a Monte Carlo search of conformation space in which allowed moves include:
- Randomization of a loop position (biased by the Ramachandran map for that residue).
- Small random perturbation of a loop position.
- Nucleation of a turn of helix (using the MakeBundle mover).
- Elongation of a helical region (with possible merger of two helical regions).
- Contraction of a helical region.
- Small random perturbation of the Crick parameters describing a given helix (using the PerturbBundle mover), to allow helices to bend and supercoil.
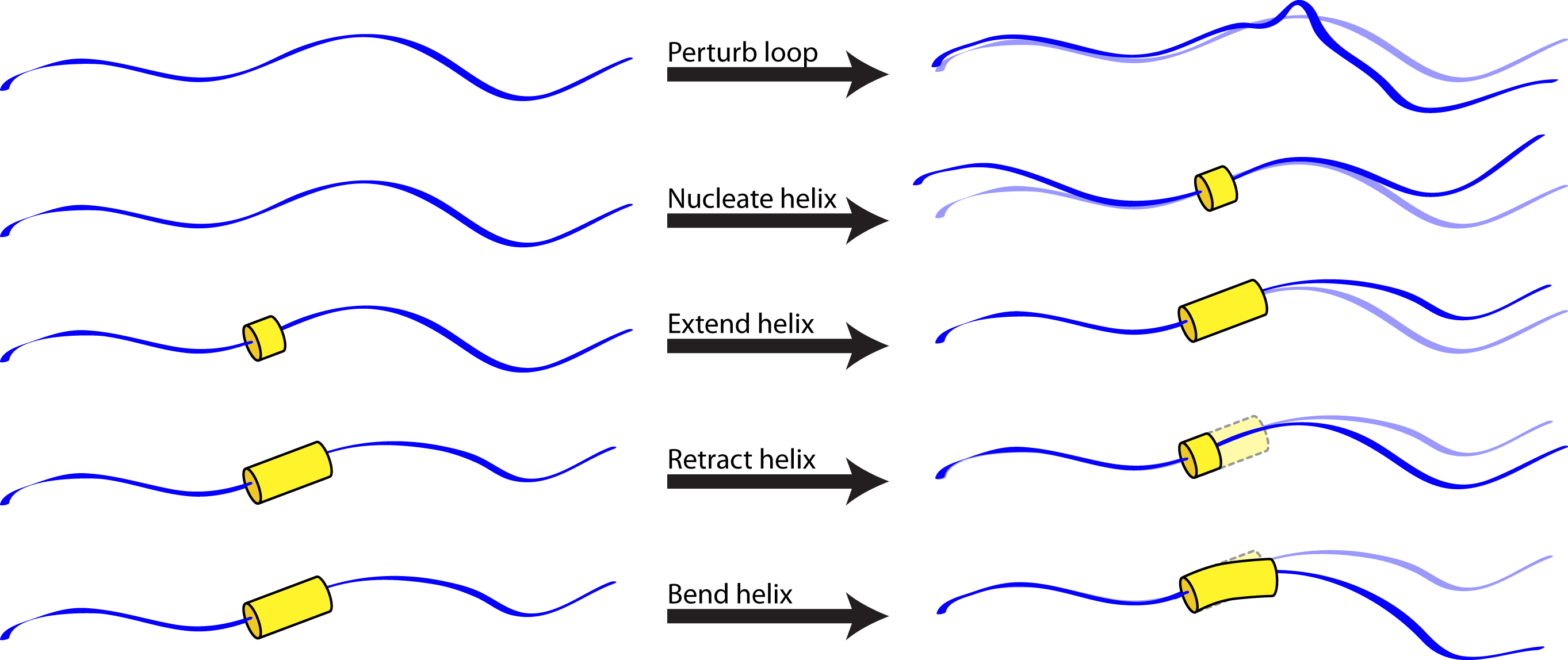
Note that, because strands are special cases of helices in which the turn per residue is about 180 degrees, this approach should be sufficiently general for any protein or heteropolymer secondary structure.
Examples
Predictions using a single core
The default build of Rosetta allows the user to launch an instance of the helical_bundle_predict application which will use one CPU core to compute a trajectory (or, if many trajectories are requested with the -nstruct option, many trajectories will be run in sequence).
Let's say that we wanted to predict the structure of a small de novo-designed 3-helix bundle miniprotein, such as PDB ID 2ND2 (designed by Chris Bahl). The following commandline would generate 500 sampled structures:
<path_to_rosetta>/Rosetta/main/source/bin/helical_bundle_predict.default.linuxgccrelease -in:file:fasta inputs/seq.txt -nstruct 500 -in:file:native inputs/2nd2.cif -in:file:fullatom -helical_bundle_predict:helix_assignment_file inputs/2nd2.helix_assignments -helical_bundle_predict:num_steps_per_simulated_annealing_round_centroid 1000 -helical_bundle_predict:num_simulated_annealing_rounds_centroid 5 -out:file:silent output.silentThis assumes that in an inputs/ subdirectory of your working directory, you have a FASTA-formatted sequence file seq.txt that contains the sequence of 2ND2, the 2ND2 native structure (2nd2.cif), and a helix assignments file (2nd2.helix_assignments) specifying the expected helical regions of the structure. See the Inputs section, below, for an example of this file for 2ND2. Note that the native structure is optional. Without it, RMSD values to the native will not be calculated. All sampled structures will be written out to output.silent.
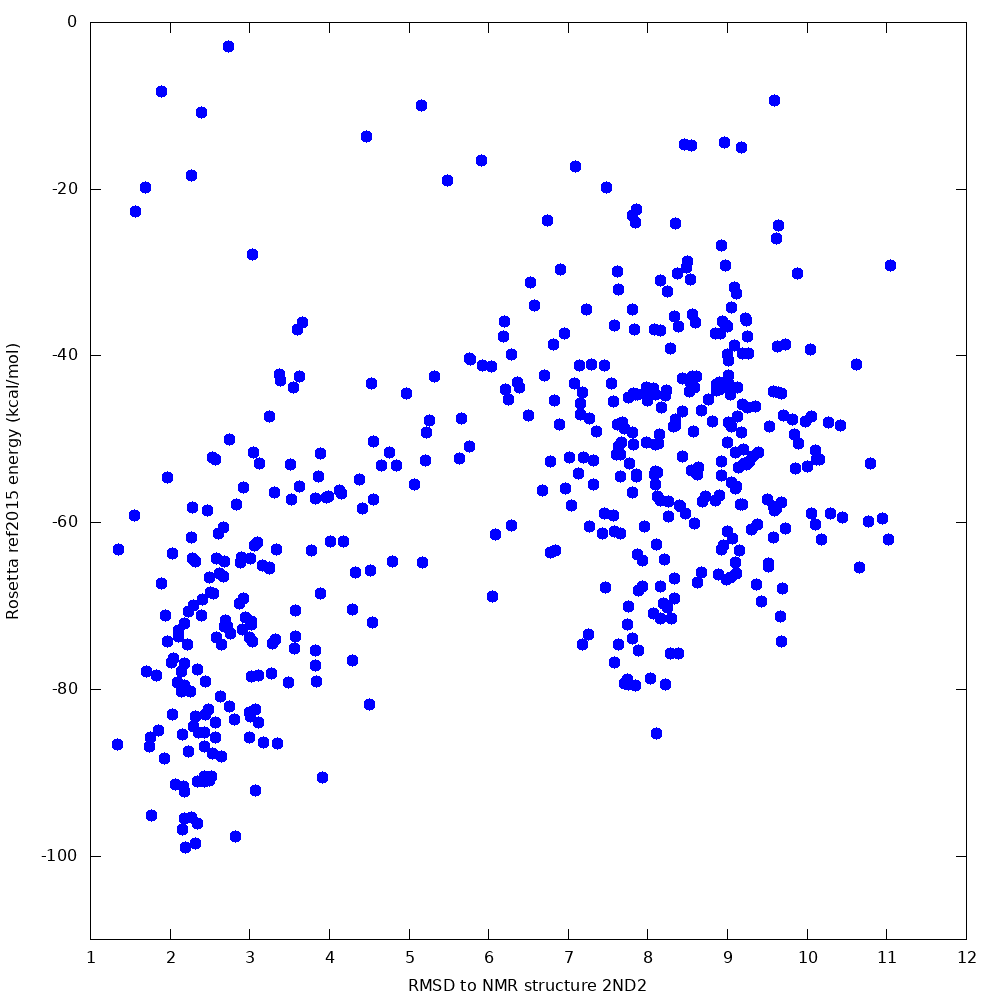
When we tested this protocol, 500 samples produced a lowest-energy sample 2.4 Angstroms from the native, with a clear funnel towards the native state:
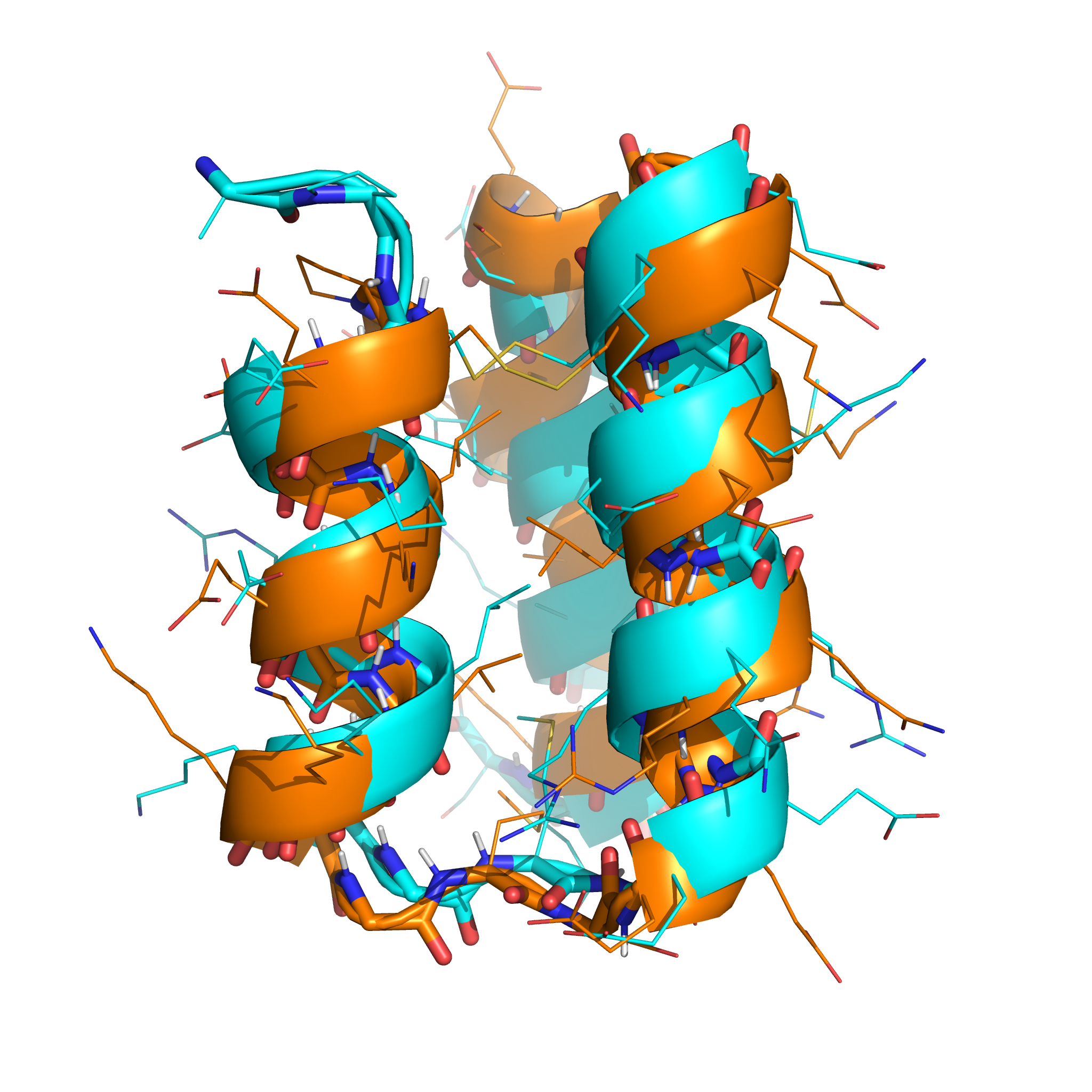
The lowest-energy sample (shown in orange, below) had topology closely resembling the native structure (shown in cyan), and the correct disulphide connectivity was found:
Predictions in parallel on a cluster
Typically, one has many cores available for prediction. The common way to take advantage of this is to run many independent, parallel instances of Rosetta -- in this case, launching as many copies of the helical_bundle_predict application as one has cores. This isn't ideal, however, for several reasons:
- Independent instances do not necessarily balance their loads well. (If the first instance finishes its work in five minutes, and the second instance still has a long list of jobs to carry out, it cannot transfer some of its work to the first. One core sits idle while another does a lot of work.)
- Independent instances have no ability to carry out analysis on the ensemble of structures generated. Any analysis must be done after-the-fact.
- Independent instances each load a separate copy of the Rosetta database into memory, potentially preventing all cores from being used on memory-limited systems.
- Independent instances all read from and write to disk independently, potentially resulting in bottlenecks as shared hardware is accessed by many independent processes on large clusters.
The MPI and multi-threaded builds address these problems.
Parallelism using Message Passing Interface (MPI) processes
With the MPI build of Rosetta (built by adding extras=mpi to the scons command during compilation), one can launch many parallel helical_bundle_predict processes. These processes can then talk to one another, allowing load-balancing, data distribution (reducing disk reads), dedicated output by one process (reducing disk writes), and data analysis over the whole pool of samples. In MPI mode, the helical_bundle_predict application establishes a hierarchy of MPI processes, in which one director processes distributes work to, and collects results from, any number of layers of intermediate manager processes, which in turn distribute work to, and collect results from, a large number of worker processes. (These layers were formerly called "emperor", "master", and "slave", and may appear as such in older versions of Rosetta.) The workers run the same structure prediction algorithm that is run by the single-process version of the application. During results collection, data analysis is also performed to (a) compute metrics like PNear, which serves as an estimate of the fraction of time a molecule spends in or near the native state (see Bhardwaj, Mulligan, Bahl et al. (2016) Nature 538(7625):329-335 and Hosseinzadeh, Bhardwaj, Mulligan et al. (2017) Science 358(6369):1461-1466), and (b) rank the samples by some metric so that only a subset might be written to disk.
To run the MPI version, follow the instructions for your cluster. Often, the mpirun command is used:
mpirun -np <# of processes> <path_to_Rosetta>/Rosetta/main/source/bin/helical_bundle_predict.mpi.linuxgccrelease @flags.txt`In the above, run options are in the flags.txt file. The -MPI_batchsize_by_level and -MPI_processes_by_level, documented in the full options list, below, must be included. As an alternative to -MPI_processes_by_level, the flag -MPI_auto_2level_distribution allows automatic setup of a two-level distribution scheme in which, given N total processes, 1 director process talks to N-1 worker processes. This flag takes no options.
Parallelism using MPI processes plus threads
Multi-threaded job distribution is also supported. To enable hybrid process/thread-based parallelism, compile with extras=cxx11threads,mpi,serialization. In the MPI/threaded build, the helica\l_bundle_predict application sends jobs down an director/manager/worker hierarchy, as before. Each worker may launch a number of threads with which to carry out samples in parallel, however (so it is important that each worker receive a batch of jobs larger than the number of worker threads that it has launched). The number of threads is controlled with the -threads_per_worker flag. Note that one thread will be used for MPI communication and job control, so launching, for example, 8 threads results in 7 worker threads.
Inputs
Helix assignment file
Helix locations and Crick parameter sampling options must be provided with an ASCII-formatted helix assignment file. This is passed to the application with the helical\_bundle\_predict:helix_assignment_file input flag. Since there are many ways of predicting secondary structure from primary sequence, and of predicting possible helix types given a polymer building block set, it is assumed that some estimates of helix locations and types will be possible to produce and provide as input. Since the algorithm stochastically nucleates, extends, and retracts helices, the helix types and ranges serve only as a guide; final helix placements may be different. Also note that strands may be specified, since as noted before they are special cases of helices. (They are still referred to as "helices" in helix assignment files.)
Helix assignment file example
The following shows an example helix assignment file, for predicting PDB ID 2ND2. The BEGIN_GLOBALS...END_GLOBALS block specifies that the helical twist (omega0) should be shared by all helices, but the bundle radius (r0) and rotation (delta_omega1) should not. Helices are not allowed to spread beyond user-defined boundaries, valid twist values range from -3 degrees to +3 degrees, valid bundle radius values range from 5 A to 8 A, and valid rotation values range from 0 to 360 degrees. All helices are standard alpha helices, defined in the alpha_helix_100.crick_params file in the Rosetta database. Monte Carlo moves have a 1% chance of nucleating a helix, a 5% chance of extending a helix, and a 3% chance of retracting a helix.
The helices are then defined in BEGIN_HELIX...END_HELIX blocks. The first ranges from position 3 through 3, the second, from position 17 through 30, and the third, from position 32 through 43. Note that the second and third helices override some of the default parameters defined in the globals section.
BEGIN_GLOBALS
COMMON_OMEGA0 TRUE
COMMON_R0 FALSE
COMMON_DELTA_OMEGA1 FALSE
CONFINE_TO_USER_DEFINED_HELICES TRUE
OMEGA0_MIN -3
OMEGA0_MAX 3
R0_MIN 5
R0_MAX 8
DELTA_OMEGA1_MIN 0
DELTA_OMEGA1_MAX 360
CRICK_PARAMS_FILE alpha_helix_100.crick_params
NUCLEATION_PROB 0.01
EXTENSION_PROB 0.05
RETRACTION_PROB 0.03
END_GLOBALS
BEGIN_HELIX
START_RES 3
END_RES 13
END_HELIX
BEGIN_HELIX
START_RES 17
END_RES 30
R0_MIN 6
R0_MAX 7
DELTA_OMEGA1_MIN 45
DELTA_OMEGA1_MAX 135
END_HELIX
BEGIN_HELIX
START_RES 32
END_RES 43
NUCLEATION_PROB 0.02
EXTENSION_PROB 0.03
RETRACTION_PROB 0.01
END_HELIXHelix assignment file format
Comments and whitespace
Comment lines may be included using a pound sign (#). Anything following a pound sign is ignored. All whitespace is treated equally within a line (i.e. preceding or trailing spaces or tabs have no significance beyond human readability).
Globals block
Global settings are defined in a block that begins with BEGIN_GLOBALS and ends with END_GLOBALS. Settings defined in the global settings may be overridden on a helix-by-helix basis in the individual helix blocks.
| Setting | Description | Example |
|---|---|---|
| COMMMON_R0 | Is the value of r0 (the radius of a helix from the bundle axis) shared by all helices, or independent? Note that this cannot be overridden on a helix-by-helix basis. | COMMON_R0 FALSE #Helices have independent r0 |
| COMMMON_OMEGA0 | Is the value of omega0 (the major helical twist) shared by all helices, or independent? Note that this cannot be overridden on a helix-by-helix basis. | COMMON_OMEGA0 TRUE #Helices share omega0 |
| COMMON_DELTA_OMEGA1 | Is the value of delta_omega1 (the rotation of a helix about its own axis) shared by all helices, or independent? (This affects helices with nonzero omega0 values, which follow a corkscrew path through space and for which different faces of the helix could point towards the center of the corkscrew.) Note that this cannot be overridden on a helix-by-helix basis. | COMMON_DELTA_OMEGA1 FALSE #Helices have independent delta_omega1 |
| CONFINE_TO_USER_DEFINED_HELICES | If true, helices cannot extend beyond user-defined helix limits. Note that this cannot be overridden on a helix-by-helix basis. | CONFINE_TO_USER_DEFINED_HELICES TRUE #User-defined limits are enforced |
| R0_MIN | The minimum radius for a helix from the superhelix axis. Can be overridden on a helix-by-helix basis. | R0_MIN 3.5 #Minimum radius of 3.5 Angstroms |
| R0_MAX | The maximum radius for a helix from the superhelix axis. Can be overridden on a helix-by-helix basis. | R0_MAX 7.5 #Maximum radius of 7.5 Angstroms |
| OMEGA0_MIN | The minimum superhelical twist. Can be overridden on a helix-by-helix basis. | OMEGA0_MIN -3.5 #Minimum superhelical twist of -3.5 degrees |
| OMEGA0_MAX | The maximum superhelical twist. Can be overridden on a helix-by-helix basis. | OMEGA0_MAX 2.5 #Maximum superhelical twist of +2.5 degrees |
| DELTA_OMEGA1_MIN | The minimum rotation of a helix about its own axis. Can be overridden on a helix-by-helix basis. | DELTA_OMEGA1_MIN 0 #Minimum axial rotation of 0 degrees |
| DELTA_OMEGA1_MAX | The maximum rotation of a helix about its own axis. Can be overridden on a helix-by-helix basis. | DELTA_OMEGA1_MAX 360 #Maximum axial rotation of 360 degrees |
| NUCLEATION_PROB | The fractional probability of nucleating a helix as a Monte Carlo move. Can be overridden on a helix-by-helix basis. | NUCLEATION_PROB 0.01 # 1% chance of nucleating a helix on any given move |
| EXTENSION_PROB | The fractional probability of extending a helix as a Monte Carlo move. Can be overridden on a helix-by-helix basis. | EXTENSION_PROB 0.05 # 5% chance of growing a helix on any given move |
| RETRACTION_PROB | The fractional probability of retracting a helix as a Monte Carlo move. Can be overridden on a helix-by-helix basis. | RETRACTION_PROB 0.03 # 3% chance of shrinking a helix on any given move |
| GLOBAL_PERTURBATION_PROB | The fractional probability of doing a global Crick parameter perturbation as a Monte Carlo move. Cannot be overridden on a helix-by-helix basis. | GLOBAL_PERTURBATION_PROB 0.02 # 2% chance of global Crick parameter perturbations |
| FRACTIONAL_PERTURBATION_MAGNITUDE | The size of perturbations to the global Crick parameters. Cannot be overridden on a helix-by-helix basis. | FRACTIONAL_PERTURBATION_MAGNITUDE 0.1 # Crick parameters are perturbed by up to 10%. |
| CRICK_PARAMS_FILE | The Crick parameters file defining the type of helix (or strand). Can be overridden on a helix-by-helix basis. | CRICK_PARAMS_FILE 14_helix.crick_params #A beta-amino acid 14-helix |
Individual helix blocks
Settings for individual helices are specified in blocks that begin with BEGIN_HELIX and end with END_HELIX.
| Setting | Description | Example |
|---|---|---|
| START_RES | The first residue of the region in which this helix might exist, based on Rosetta numbering (starting at 1). | START_RES 5 #This helix starts somewhere on or after residue 5. |
| END_RES | The last residue of the region in which this helix might exist, based on Rosetta numbering (starting at 1). | END_RES 15 #This helix ends somewhere before or on residue 15. |
| R0_MIN | Minimum value for radius from supercoiling axis. Overrides any global setting. Cannot be used with COMMON_R0. | R0_MIN 3.65 # This helix has a minimum radius from the supercoiling axis of 3.65 Angstroms. |
| R0_MAX | Maximum value for radius from supercoiling axis. Overrides any global setting. Cannot be used with COMMON_R0. | R0_MAX 8.0 # This helix has a maximum radius from the supercoiling axis of 8.0 Angstroms. |
| OMEGA0_MIN | Minimum value for supercoiling twist. Overrides any global setting. Cannot be used with COMMON_OMEGA0. | OMEGA0_MIN -1.0 # This helix has a minimum supercoil of -1.0 degrees. |
| OMEGA0_MAX | Maximum value for supercoiling twist. Overrides any global setting. Cannot be used with COMMON_OMEGA0. | OMEGA0_MAX +1.0 # This helix has a maximum supercoil of +1.0 degrees. |
| DELTA_OMEGA1_MIN | Minimum value for the rotation of a helix about its own axis. Overrides any global setting. Cannot be used with COMMON_DELTA_OMEGA1. | DELTA_OMEGA1_MIN 45 # This helix has a minimum rotation about its own axis of 45 degrees. |
| DELTA_OMEGA1_MAX | Maximum value for the rotation of a helix about its own axis. Overrides any global setting. Cannot be used with COMMON_DELTA_OMEGA1. | DELTA_OMEGA1_MAX 135 # This helix has a minimum rotation about its own axis of 135 degrees. |
| NUCLEATION_PROB | Probability of nucleating a helix in this region on any given Monte Carlo move. Overrides any global setting. | NUCLEATION_PROB 0.08 # This region has a nucleation probability of 8%. |
| EXTENSION_PROB | Probability of extending a helix in this region on any given Monte Carlo move. Overrides any global setting. | EXTENSION_PROB 0.02 # This region has a helix-growing probability of 2%. |
| RETRACTION_PROB | Probability of retracting a helix in this region on any given Monte Carlo move. Overrides any global setting. | RETRACTION_PROB 0.02 # This region has a helix-shrinking probability of 2%. |
| CRICK_PARAMS_FILE | The helix type. Overrides any global setting. | CRICK_PARAMS_FILE beta_strand.crick_params #This is a beta strand. |
PsiPred ss2 secondary structure prediction file
In addition to a helix assignment file, the helical_bundle_predict application can accept a PsiPred ss2 file. This has rows for each position in the protein, with coil, helix, and strand probabilities listed in the fourth, fifth, and sixth columns in each row. Any position with a helix probability over a threshold is assigned to be in an alpha helix, and any position with a strand probability over a threshold is assigned to be in a beta strand. Note that this is only useful for assigning canonical secondary structures to canonical amino acids.
To use a PsiPred ss2 file with this application, provide it with the -helical_bundle_predict:psipred_file commandline flag. The thresholds for alpha helices and beta strands can be set with the -helical_bundle_predict:psipred_alpha_helix_prob_cutoff and -helical_bundle_predict:psipred_beta_strand_prob_cutoff flags. Note that this can be used in addition to a helix assignment file. Since the helix assignment file is the only way to provide global options, it is generally recommended to include one.
Full options list
| Option | Default Setting | Type | Description |
|---|---|---|---|
| in: file: fasta |
File | Fasta-formatted sequence file. | |
| helical_bundle_predict: sequence_file |
File | Filename of a file specfying the sequence, as a series of whitespace-separated full residue names (e.g. ALA LYS DARG DPRO HYP). Required input for the simple_cycpep_predict app unless -in:file:fasta is provided. | |
| in: file: native |
File | Native PDB filename. | |
| out: nstruct |
1 | Int | Number of structures to generate. (Number of structure prediction attempts) |
| helical_bundle_predict: helix_assignment_file |
File | A file containing information about the helix types and helical regions within a helical bundle. | |
| helical_bundle_predict: psipred_file |
File | A file produced by PsiPred, with columns indicating position, alpha helix probability, beta strand probability, and random coil probability. An alternative to helix_assignment_file for canonical amino acid predictions. | |
| helical_bundle_predict: psipred_alpha_helix_prob_cutoff |
0.75 | Real | The probability cutoff above which a position is assigned to be alpha helical when using a PsiPred file. Default 0.75. |
| helical_bundle_predict: psipred_beta_strand_prob_cutoff |
0.75 | Real | The probability cutoff above which a position is assigned to be in a beta strand when using a PsiPred file. Default 0.75. |
| helical_bundle_predict: num_steps_per_simulated_annealing_round_centroid |
1000 | Int | Number of steps in each round of simulated annealing in centroid mode. |
| helical_bundle_predict: num_simulated_annealing_rounds_centroid |
3 | Int | Number of rounds of simulated annealing in centroid mode. |
| helical_bundle_predict: centroid_max_temperature |
50 | Real | The maximum temperature during simulated annealing rounds in centroid mode. |
| helical_bundle_predict: centroid_min_temperature |
0.62 | Real | The minimum temperature during simulated annealing rounds in centroid mode. |
| helical_bundle_predict: do_final_fullatom_refinement |
true | Bool | If true, the initial centroid model is converted to a full-atom model and relaxed with the FastRelax protocol. Other refinement steps, such as finding disulfides, may also be carried out. True by default. |
| helical_bundle_predict: fast_relax_rounds |
3 | Int | The number of rounds of FastRelax that will be applied. Does nothing if do_final_fullatom_refinement is false. Set to 3 by default. |
| helical_bundle_predict: find_disulfides |
true | Bool | If true, the full-atom refinement steps include trying disulfide permutations. Does nothing if do_final_fullatom_refinement is false. True by default. |
| helical_bundle_predict: ignore_native_residues_in_rmsd |
(Empty list) | Int vector | A whitespace-separated list of residues in the pose provided with -in:file:native which should NOT be used in calculating the RMSD. The alignment will skip these residues. For example, if the generated structures are 10 residues long, the native is 11, and residue 5 is skipped, then residues 1-4 will be aligned to residues 1-4 of the generated structures, and residues 6-11 of the native wil be aligned to resiudues 5-10 of the generated stuctures. |
| helical_bundle_predict: ignore_prediction_residues_in_rmsd |
(Empty list) | Int vector | A whitespace-separated list of residues in the generated poses that will be ignored when aligning to the pose provided with -in:file:native. These residues are skipped in the alignment -- so if, for example, residues 3 and 4 are specified, and the generated structures are 12 residues long and the native structure is 10 residues long, residues 1-2 of both structures will be aligned, and residues 5-12 of the generated structures will be aligned to residues 3-10 of the native. |
| cyclic_peptide: MPI_processes_by_level |
Int vect | The number of processes at each level of the parallel communications hierarchy, used only by the MPI version. For example, '1 10 100' would mean that one director would talk to 10 managers, which would talk to 100 workers (implying that each manager is assigned 10 workers). Similarly, '1 100' would mean that one manager would talk directly to 100 workers. Required for the MPI version. | |
| cyclic_peptide: MPI_auto_2level_distribution |
false | Bool | If true, will automatically set up job distribution with one director talking to N-1 workers, where N is the total number of processes. Cannot be used with -MPI_processes_by_level. Default false. |
| cyclic_peptide: MPI_batchsize_by_level |
Int vect | The number of jobs sent at a time by each communication level to its children. Given N levels, N-1 values must be specified. For example, given 3 communications levels, '100 10' would mean that the director sends 100 jobs at a time to each manager, which sends 10 jobs at a time to each worker. Must be specified for the helical_bundle_predict application in MPI mode. | |
| cyclic_peptide: MPI_sort_by |
"energy" | String | The MPI version of the helical_bundle_predict app has the option of writing out the top N% of solutions. This determines the sort metric. |
| cyclic_peptide: MPI_choose_highest |
false | Bool | When outputing the top N% of solutions, should I choose the ones with the highest score for the metric chosen (energy, rmsd, hbonds, etc.), or the ones with the lowest? False by default (chooses lowest). |
| cyclic_peptide: MPI_output_fraction |
1 | Real | The fraction of total structures that will be written out. This is used in conjunction with 'MPI_sort_by' to output the top N% of job outputs. For example, '-MPI_output_fraction 0.05 -MPI_sort_by rmsd' means that the 5% of structures with the lowest RMSD values will be written out. |
| cyclic_peptide: MPI_stop_after_time |
I | If this option is used, the director node will send a stop signal after an elapsed period of time, given in seconds. Worker jobs currently running will continue, but intermediate managers will not assign any more work. Useful on HPC clusters with time limits, to ensure that jobs completed are collected at the end. Unused if not specified. | |
| cyclic_peptide: MPI_pnear_lambda |
0.5 | Real | In MPI mode, a goodness-of-funnel metric is automatically calculated at the end (PNear). This value may be thought of as the probability, from 0 to 1, of the peptide being in the native conformation at any given time. The parameter lambda controls the breadth of the Gaussian (in RMSD units -- Angstroms) that is used to determine the extent to which a state is native-like. Default 0.5 A. |
| cyclic_peptide: MPI_pnear_kbt |
1 | Real | In MPI mode, a goodness-of-funnel metric is automatically calculated at the end (PNear). This value may be thought of as the probability, from 0 to 1, of the peptide being in the native conformation at any given time. The parameter kbt is the Boltzmann temperature that determines the extent to which higher energy states are likely to be sampled. Default 1.0 kcal/mol. |
| cyclic_peptide: compute_rmsd_to_lowest |
false | Bool | If true the RMSD to the top structure (by whatever ranking) is computed in addition to the RMSD to a user-supplied native (if a native structure is provided). False by default. Only used in MPI version of Rosetta. |
| cyclic_peptide: compute_pnear_to_this_fract |
0.0 | Real | If this option is provided, PNear will be computed to each of the lowest-energy samples found, for the specified lowest-energy fraction. (Setting this to 0.02 would compute PNear to each of the lowest-energy 2% of samples, for example). Not used by default.) Only used in MPI version of Rosetta. |
| cyclic_peptide: threads_per_worker |
1 | I | In the multi-threaded MPI compilation, this is the number of threads to launch per worker process. Note that director and manager-layer processes do not launch threads. A value of 1 (the default) means that only standard hierarchical process-based parallelism will be used. In non-MPI or non-threaded compilations, this option is unused. |
Code organization
Base protocol
The application is located in src/apps/public/helical_bundle/helical_bundle_predict. The protocol that it runs is located in src/protocols/helical_bundle_predict, with classes defined in the protocols::helical_bundle_predict namespace.
The main protocol is defined in the protocols::helical_bundle_predict::HelicalBundlePredictApplication class, in src/protocols/helical_bundle_predict/HelicalBundlePredictApplication.hh. The MPI/multi-threaded variant (which calls the HelicalBundlePredictApplication for individual prediction trajectories) is defined in the protocols::helical_bundle_predict::HelicalBundlePredictApplication_MPI class, which derives from the protocols::cyclic_peptide_predict::HierarchicalHybridJDApplication base class and uses the same hierarchical MPI/multi-threaded job distribution and results collection system as the simple_cycpep_predict application.
Support classes
A number of support classes are also defined in the protocols::helical_bundle_predict namespace, in header files corresponding to the class name. These include:
-
HBPHelixAssignments-- Stores the helix assignments for a structure prediction task. Separate instances of this class are used both for the user-defined helix assignments and for the current helix assignments at a given stage of a trajectory. Functions to read assignments from disk are included in this class. -
HBP_MoveGenerator-- An abstract base class for generating ParsedProtocols representing moves in a Monte Carlo trajectory. -
HBP_HelixCoilMoveGenerator-- Generates a ParsedProtocol for a single move in the centroid-mode Monte Carlo trajectory, based on the current state of the pose. The move is composed of a random mix of loop perturbations, helix nucleations, helix elongations, helix contractions, and helix parameter perturbations (helix bending moves) with probabilities set by the user. Derived fromHBP_MoveGenerator. -
HBP_FinalFullatomRefinementMoveGenerator-- Generates a ParsedProtocol for final full-atom refinement of a structure, at the end of a centroid-mode conformational sampling trajectory. Derived fromHBP_MoveGenerator. -
HBP_TemperatureScheduleGenerator-- An abstract base class for classes that generate the temperature schedule for a Simulated Annealing trajectory. -
HBP_SigmoidalTemperatureScheduleGenerator-- A class for generating a sigmoidal temperature schedule for a simulated annealing trajectory, derived fromHBP_TemperatureScheduleGenerator.
Current limitations
This application was developed to allow non-canonical heteropolymer structure prediction. At the present time, it has only been tested on proteins. A current limitation is that initial sampling is carried out in centroid mode, requiring a centroid-mode scoring function that is compatible with the polymer building blocks used. Since Rosetta's current centroid scoring functions are hard-coded to be specific for canonical amino acids, they cannot be used for non-canonical heteropolymers.
Development of a new, general centroid scoring function is ongoing.
See also
- Rosetta ab initio application -- Fragment-based protein structure prediction.
- Rosetta simple_cycpep_predict application -- Structure prediction of macrocycles built from canonical or non-canonical building-blocks.
- Generalized kinematic closure -- A mover to sample conformations of a closed chain of atoms, without fragments.
- MakeBundle mover -- A mover that generates a coiled-coil protein or heteropolymer parametrically, using the Crick equations.
- PerturbBundle mover -- A mover that alters Crick parameter values to perturb the conformation of a coiled-coil.
- BundleGridSampler mover -- A mover that grid-samples Crick parameter space to identify favourable coiled-coil conformations.