Membrane Energy Functions (RosettaMP)
Overview
The current implementation of RosettaMP uses the same science (i.e. score terms) of the original RosettaMembrane implementation. To increase the flexibility & user-friendliness, we implemented a new interface for these terms. The science & interface for the low- and high-resolution scoring functions are described below.
Low Resolution Scoring Function
The Rosetta low resolution (centroid-based) energy function for membrane proteins was developed in 2006 with the Membrane Ab Initio folding protocol (Yarov-Yaravoy et al. 2006). It was derived from 28 alpha helical membrane proteins and is knowledge-based.
Membrane Representation
The low resolution energy function divides the membrane bilayer into two or five layers describing various regions of the membrane environment. Scoring per-residue, residue-residue and packing interactions depends upon position in a specific layer. The five-layer representation is described below:
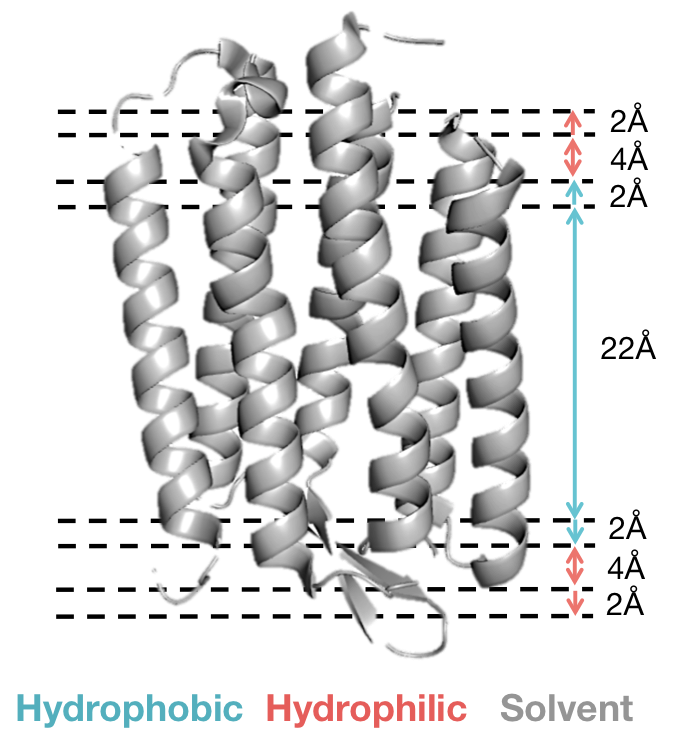
Scoring Function
The energy function includes six terms: a residue-environment energy, residue-residue energy, packing density, termini penalty, non-helix penalty, and TMprojection penalty.
For a detailed description of all score terms, their functional form and their weights for specific applications, please see Alford, Koehler Leman et al.
Individual energy terms
Terms, corresponding energy methods, and usage are described below. The score name is the name of the term you would use when setting up a weights file.
Membrane Residue-Environment Term
Score Name: MPEnv. Score per-residue interactions with the membrane environment using a 5 layer membrane (Layers: inner hydrophobic, outer hydrophobic, interface, polar, water). Energy Method type: One body context-dependent. For Developers - code in core/scoring/membrane/MPEnvEnergy.hh.
Membrane Residue-Residue Pair Term
Score Name: MPPair. Score residue-residue interactions in the membrane environment using a 2 layer membrane (Layers: hydrophobic, polar). Energy Method Type: Two-body context dependent. For Developers - code in core/scoring/membrane/MPPairEnergy.hh
Membrane Packing Density Term
Score Name: MPCBeta. Score packing-density in the membrane based on the number of helices. Energy Method Type: One Body context dependent. For Developers - code in core/scoring/membrane/MPCbetaEnergy.hh
Membrane Termini Penalty
Score Name: MPTermini. Penalty for termini residues (N- or C- termini) in the membrane. Energy Method Type: Whole structure energy. For Developers - code in /core/scoring/membrane/MPTerminiPenalty.hh
Membrane TM Projection Penalty
Score Name: MPTMProj. Penalty for helices longer than thickness of the membrane. Energy Method type: Whole Structure Energy. For Developers - code in core/scoring/membrane/MPTMProjPenalty.hh
Membrane Non Helix Penalty
Score Name: MPNonHelix. Penalty for non helix residues in the membrane. Uses Secondary structure in Rosetta (or provided). Energy Method Type: One body context independent energy. For Developers - code in core/scoring/membrane/MPNonHelixPenalty.hh
Score Options
| Option Name | Description | Type |
|---|---|---|
| score:find_neighbors_3dgrid | Use 3D lookup table in neighbor calculations | boolean |
| membrane:no_interpolate_Mpair | switch off interpolation between 2 layers when a pair of residues is on the boundary | boolean |
High-Resolution Scoring Function
The Rosetta high resolution (all atom) energy function was developed in 2007 with the high-resolution ab initio and refinement protocols (Barth et al. 2007) and is based on the Implicit Membrane Model by Lazaridis (2003).
Membrane Representation
The high-resolution energy function represents the membrane bilyaer as a hydrophobic slab of fixed thickness, T (where the default thickness is 30Ang). This gives a hydrophobic core of 18A with transition regions of 6A on either side. This membrane representation is illustrated below:
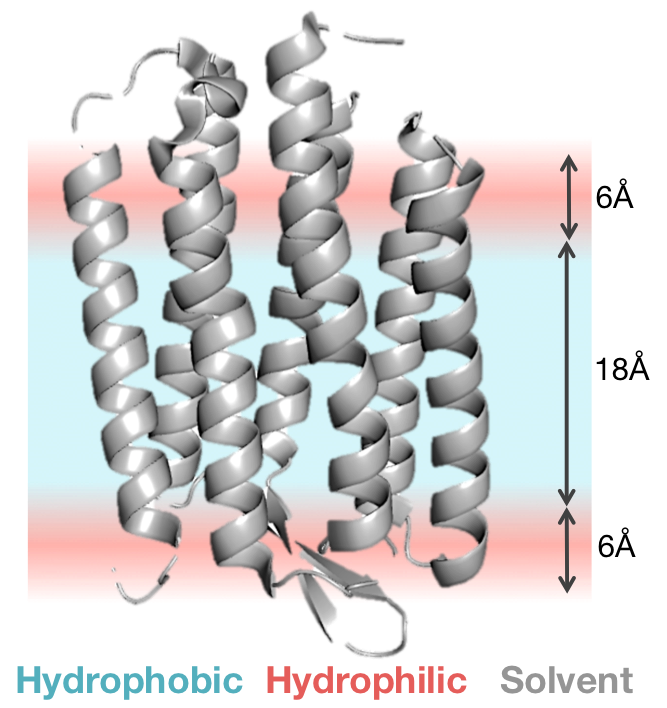
Scoring function
The high-resolution score function combines terms from the score12 energy function with terms for membrane solvation, environment and a correction for strength of hydrogen bonds in the membrane.
For a detailed description of all score terms, their functional form and their weights for specific applications, please see Alford, Koehler Leman et al.
Individual Energy Terms
Membrane Solvation
Score Name: fa_mpsolv. Score a pair of residues given their depth in the membrane bilayer
Membrane environment
Score Name: fa_mpenv. Score a single residue given its depth in the membrane bilayer
Score Options
| Option Name | Description | Type |
|---|---|---|
| mp:scoring:hbond | Hydrogen bonding energy correction for membrane proteins. Default = false. | boolean |
References
- Alford RF, Koehler Leman J, Weitzner BD, Duran A, Tiley DC, Gray JJ (2015). An integrated framework advancing membrane protein modeling and design. PLoS Comput. Biol. - In Press - Rosetta Revision #57518
- Yarov-Yarovoy V, Schonbrun J, Baker D. (2006 Mar 1) Multipass membrane protein structure prediction using Rosetta. Proteins. 62(4):1010-25.
- Barth P, Schonbrun J, Baker D. (2007 Oct 2) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 104(40):15682-7.
- Barth P, Wallner B, Baker D. (2009 Feb 3) Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U S A. 106(5):1409-14.
Contact
- Rebecca Alford (rfalford12@gmail.com)
- Julia Koehler Leman (julia.koehler1982@gmail.com)
- Corresponding PI: Jeffrey J. Gray (jgray@jhu.edu)