Membrane Representation - The MEM Residue (RosettaMP)
Description
In RosettaMP, the position and orientation of the membrane is represented by a special residue, of type MEM, typically found at the end of the pose sequence. It stores the center point of the membrane, the membrane normal vector, and the thickness. The thickness value stored in MEM is half of the "true" membrane thickness, since the membrane is currently regarded as symmetric around z = 0 being at the center of the membrane.
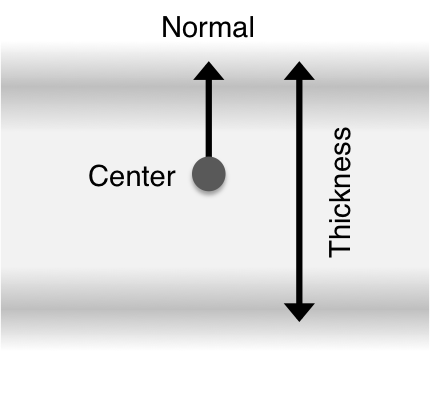
The MembraneResidue is automatically printed to the PDB as the MEM residue in a HETATOM record. This allows for easy visualization of the membrane in Pymol. The normal vector is stored as an "atom" and is of unit length. Be aware that the normal is a vector in the coordinate system, but stored as a point in the PDB file. For easy visualization, the normal can be scaled to the thickness using the flag -mp:output:normalize_to_thk
HETATM 1323 THKN MEM C 81 1.000 0.000 0.000 1.00 0.00 X
HETATM 1324 CNTR MEM C 81 -1.425 -1.441 0.354 1.00 0.00 X
HETATM 1325 NORM MEM C 81 0.065 -0.249 0.966 1.00 0.00 X In the above example the normal vector is close to the direction of the positive z-coordinate, even though the exact normal vector would need to be calculated by (normal 'point' minus center point).
The thickness is stored on the x-axis because a residue Stub (i.e. coordinate system) can only be created from three atoms that are not coplanar. For visualization purposes the thickness atom is not really needed, but the thickness value that the Membrane Framework used to create that decoy is stored (and can be therefore be easily accessed) as the THKN atom in the PDB file.
The coordinates of the MembraneResidue are automatically updated during a simulation. An existing MembraneResidue from a previously run protocol can be read in for a new protocol using the option -mp::setup::membrane_rsd <membrane residue number>.
The MembraneResidue is connected to the pose by a jump in the FoldTree, anchored at the first residue of the protein, but written out into the PDB file as the last residue after the protein (HETATOM record of MEM). Depending on where the root of the FoldTree is, either the membrane or the protein is flexible or fixed: (1) If the MembraneResidue is at the root of the FoldTree, it remains fixed while the protein moves flexibly in this coordinate system. (2) If one of the protein residues is at the root of the FoldTree, the MembraneResidue (and therefore the membrane) moves flexibly in the coordinate system of the pose.
Flags
The membrane residue (MEM HETATM lines in the PDB) is automatically detected by the AddMembraneMover. If for whatever reason (since protocols are still in development) more than one residue is present, the user can specify which residue should be considered by providing the flag
-mp::setup::membrane_rsd <membrane residue number> for reading in the MEM residue.
References
Alford RF, Koehler Leman J, Weitzner BD, Duran A, Tiley DC, Gray JJ (2015). An integrated framework advancing membrane protein modeling and design. PLoS Comput. Biol. - In Press
Contact
- Rebecca Alford (rfalford12@gmail.com)
- Julia Koehler Leman (julia.koehler1982@gmail.com)
- Corresponding PI: Jeffrey J. Gray (jgray@jhu.edu)