Antibody Design Analysis
Antibody design analysis consists of two parts: analyze_antibody_design_strategy.py and compare_antibody_design_strategies.py
These will eventually be moved to Rosetta/tools/antibody/analysis
Setup
Requirements include:
- PyRosetta
- Biopython
- Compiled Rosetta
- Rosetta Tools
Clustal Omega
All Decoys should be uniquely named
A basic understanding of how to edit your shell profile (if using zsh, this can be edited via $HOME/.zshrc. If you are using Bash on linux, the profile is at $HOME/.bashrc if mac, then $HOME/.bash_profile). Remember, no spaces before or after any equal signs. More information on this can be found throughout the web. this for example.
PyRosetta Setup
Download PyRosetta from https://www.pyrosetta.org or compile it from C++ Rosetta. Add the PyRosetta directory to your path by either running SetPyRosettaEnvironment.sh or by adding this to your bashrc/zshrc or whatever shell profile you are using so that SetPyRosettaEnvironment is run every time you open a new shell (source path/to/SetPyRosettaEnvironment.sh). This is preferred. This will enable import of PyRosetta and associated PyRosetta Toolkit modules located in pyrosetta/apps/pyrosetta_toolkit.
BioPython Setup
Biopython is used to load structures quicker then PyRosetta to output combined FASTA files for running Clustal Omega. PyRosetta is great, but memory and speed are important when you have hundreds or thousands of structures.
To install Biopython, run setup_biopython.sh located in the directory containing the analysis scripts
Rosetta Setup
Rosetta should be compiled. Rosetta will be used to run the FeaturesReporters through RosettaScripts. The Rosetta/main/bin directory should be added to your path in your shell profile, and you should set the Rosetta database. This will make it so that you do not need to give the database path while running Rosetta. export ROSETTA3_DB=path/to/Rosetta/main/database
Rosetta Tools
The Rosetta Tools repository should be cloned (for developers) or downloaded with the rest of Rosetta.
The path to this repository should be added to your pythonpath in your shell profile. For example, I add this line to my zshrc file: export PYTHONPATH=$PYTHONPATH:/Path/to/Rosetta/tools You will need to checkout the antibody_tools branch until I merge this into git.
Clustal Omega
The last thing you need to run all this is clustal Omega. This is to create alignments between you top designs, and between your strategies. See http://www.clustal.org/omega/ for download instructions. The clustal binary executable should be renamed clustal_omega, and the directory in which it resides should be added to your path, for example: export PATH=$PATH:/path/to/Rosetta/bin
Analyzing Antibody Design Strategies
This script, analyze_antibody_design_strategy.py analyzes one strategy at a time. You should run it from the directory it is in. Use ./analyze_antibody_design_strategy.py --help to get an idea of the options available. Generally, it should only presently be used for the creation of the FeaturesReporter databases. These will have an analysis of DGs, energies, hbonds, cdr lengths, etc for each PDB in the strategy. It is useful to run the code via a shell script for all the strategies you have available, as these can be backgrounded to analyze multiple strategy runs simultaneously.
Required Options:
Input
Input can either be a txt file with a list of PDBs, or a directory of PDBs.
--PDBLIST path/to/pdblist- This PDBLIST can either have full or relative paths to the PDBs. If relative, then the next option should be set
--indir path/to/pdbs- This Should be set if you want to parse a single directory (non-recursively) or you have relative paths in your PDBLIST
--rosetta_extension linuxclangrelease for example- - This is the Rosetta extension that will be used to call RosettaScripts to create the features databases. Generally, it is in this format: system - compiler - mode. System for mac is macos. Default for compiler is gcc, but may be clang on mac systems. Release is much quicker then debug mode. Why debug mode is the default mode is beyond me.
Output
--outdir- The relative or full output directory. If not created, will create it. Databases will be placed in outdir/databases
--out_name- The output name of the resultant database.
- The output name of the resultant database.
--out_db_batch- The batch name of the experiment that will go into the resultant database.
--score_weights talaris2013- - Optionally set a weights file to use when adding scoring features or rescoring. If you have a patch, you will need to create a full weights file instead and put it in the rosetta database.
Analysis Options
You can run all analysis by passing the --do_all option, however, this is not recommend as rescoring via PyRosetta takes a very long time. We will use the scores generated and output into the features database, as well as output tops, write pymol sessions, etc. during our comparison. It is recommended to run the antibody_features reporters on all of your models, but you can run them on only top scoring. See -help for more scoring options. This is not recommended, as you will need to rescore all the models before the run.
Recommended:
-
--do_run_antibody_features_all- Pass to enable. Run the AntibodyFeatures to create databases for the strategy on all PDBs
Not Recommended:
--do_run_antibody_features_top- Pass to enable. Run the AntibodyFeatures to create databases for the strategy on the top x PDBs. If not rescored, will rescore.
--do_run_cluster_features_all- Pass to enable. Run only the ClusterFeatures reporter on all PDBs in list or directory
--do_run_cluster_features_top- - Pass to enable. Run the ClusterFeatures reporter on the top x PDBs of the strategy. Rescore PDB list if necessary.
Comparing Antibody Design Strategies
This application compares and outputs alignments, scores, and sessions from pre-analyzed antibody designs. Each strategy should have a features database associated with it.
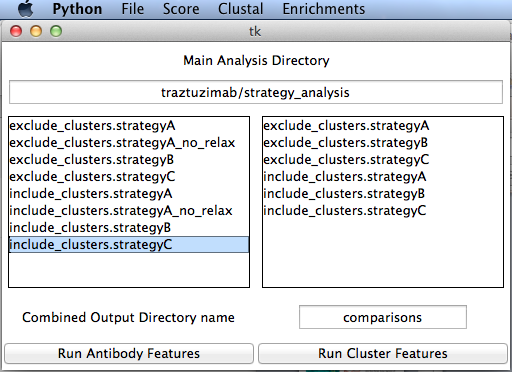
Launch in the directory where the program exists, optionally passing the full or relative path to the main analysis directory. The program will look for a directory called /databases within that directory. This is created using the analysis script. If no path is given, you can set the path in the File menu.
./compare_antibody_design_strategies.py traztuzimab/strategy_analysis
Double click a strategy on the left to move it to the right box to analyze it. Double click a strategy from the right to remove it from the list.
Select Run Antibody Features to run the set of R scripts to plot all available data and compare strategies. This may take a fairly long time. Cluster Features are used for recovery if a reference database is set in the file menu. It is used mainly for benchmarking.
- Caveats: Hbond calculations can be extremely slow. If they take too long, edit the file:
Rosetta/tools/antibody/analysis/antibody_features.json. This file has the list of all the R scripts we will run for the analysis. Remove any hbond scripts you see, especially intra-cdr hbonds. Make sure to be in a different branch if you are a developer. We will add an option to include or exclude hydrogen bond calculations while running the features comparisons shortly.
- Caveats: Hbond calculations can be extremely slow. If they take too long, edit the file:
File
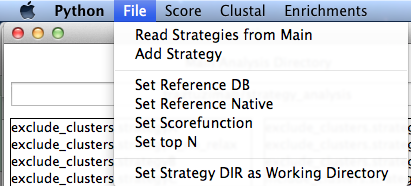
Here you can set a new analysis directory or add a new strategy. If you have a reference PDB, it is useful to set it here. If using a different scorefunction than talaris2013, it should be set here as well. It is used to search for the database of interest. You can also set the Top N. This is used throughout the program and the default is 10. Additionally, if you have a reference database (perhaps 100 relax models of the native decoy analyzed into a features database using analyze_antibody_design_strategies, it will set here and labeled as ref)
Score
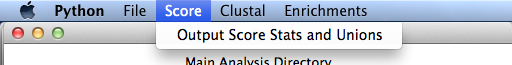
- Output Score Stats
- Here we analyze the scores of all the decoys in the strategies selected and output stats including means and standard deviations of those scores (total, dG, and dSASA) as well as the top scoring - Both combined (over all the strategies) and individually. Eventually, we will be able to set individual score terms to be included in this list. I hope to get to this soon. Optionally, you can copy the top scoring into a new directory and make a pymol session of them which includes any native antibody set.
Copy All Decoys
- - This will copy all the decoys into a new directory, with names reflecting the scores. In addition, you can output the top scoring as a pymol session just like the other option.
Clustal
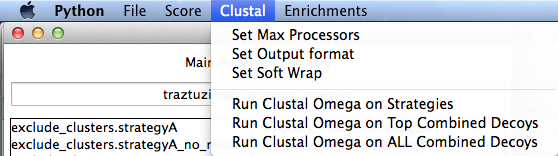
This runs Clustal Omega. You can run them on the top of each score type (currently total_score, dG, dSASA), or ALL of your decoys. You will get a chance to add additional clustal options when you select the button to run.
- Set Max Processors
- Set the max number of processors to use for Clustal Omega.
Set Output Format
- Clustal can output in many formats. Default is clustal.
- Clustal can output in many formats. Default is clustal.
Set Soft Wrap
- - You can set this large or small, and will tell clustal to only use a specific amount of spaces when outputting. Default is 100.
Eventually, we will output Clustal alignments on only the CDRs of interest and or the framework.
Enrichments
- Recovery
- Output CDR Length and Cluster recoveries to the set reference antibody for combined and separate strategies.
Alignments
- Output CDR Length and Cluster alignments for combined and separate strategies.
Enrichment
- - Not currently implemented. Will eventually give enrichment information of each CDR length and cluster. Perhaps even positional information derived from clustal omega and multiple CDR statistics.
Filters
These are accessed by the File menu. You can set dG, dSASA, and total score filters that will be used across the whole program (except for the Antibody Features R scripts). You can also set custom filters that use sqlite3 syntax. This is useful to exclude certain lengths or a minimum number of antigen contacts.
See Also
- General Antibody Options and Tips
- PyRosetta: The PyRosetta wiki page
- PyRosetta website (external)
-
Antibody Applications: Homepage for antibody applications
- Antibody protocol: The main antibody modeling application
- Antibody Python script: Setup script for this application
-
Grafting CDR loops: Graft antibody CDR templates on the framework template to create a rough antibody model.
- Modeling CDR H3: Determine antibody structures by combining VL-VH docking and H3 loop modeling.
- Camelid antibody docking: Dock camelid antibodies to their antigens.
- SnugDock: Paratope structure optimization during antibody-antigen docking
- Antibody Design Strategy Analysis: A PyRosetta-based tool to analyze and/or compare antibody design strategies.
-
CDR Cluster Identification: An application that matches each CDR of an antibody to North/Dunbrack CDR clusters based on the lowest dihedral distance to each cluster center.
- CDR Cluster Constrained Relax: An application to relax CDRs using circular harmonic constraints based on identified CDR clusters.
- Application Documentation: Application documentation home page
- Running Rosetta with options: Instructions for running Rosetta executables.
- Analyzing Results: Tips for analyzing results generated using Rosetta
- Rosetta on different scales: Guidelines for how to scale your Rosetta runs