Getting Started Modeling Membrane Proteins with RosettaMP
Overview
For modeling membrane proteins, Rosetta relies on a framework called RosettaMP for representing the physical and chemical characteristics of the membrane environment. RosettaMP was developed by Rebecca Alford and Julia Koehler Leman at the GrayLab at Johns Hopkins in 2015 and is described in Alford, Koehler Leman et al. 2015
History
In 2006 and 2007, the first membrane protein modeling applications (called RosettaMembrane) were added to Rosetta by Valdimir Yarov-Yarovoy, Patrick Barth and Bjoern Wallner. These applications introduced a low- and high- resolution scoring function and laid the groundwork for ab initio folding in the membrane bilayer. In order to advance membrane protein modeling in Rosetta, we needed a framework fully integrated with Rosetta 3.
In 2014, we decided to create a completely new machinery for membrane modeling to (1) greatly facilitate new protocol development for membrane proteins, (2) connect this code to newer concepts such as FoldTree, Movers, Graph, ScoreFunctionFactory, etc, (3) improve user-friendliness, (4) make protocols available for RosettaScripts, PyRosetta, and commandline. For more detail please see Alford, Koehler Leman et al.
Elements of RosettaMP
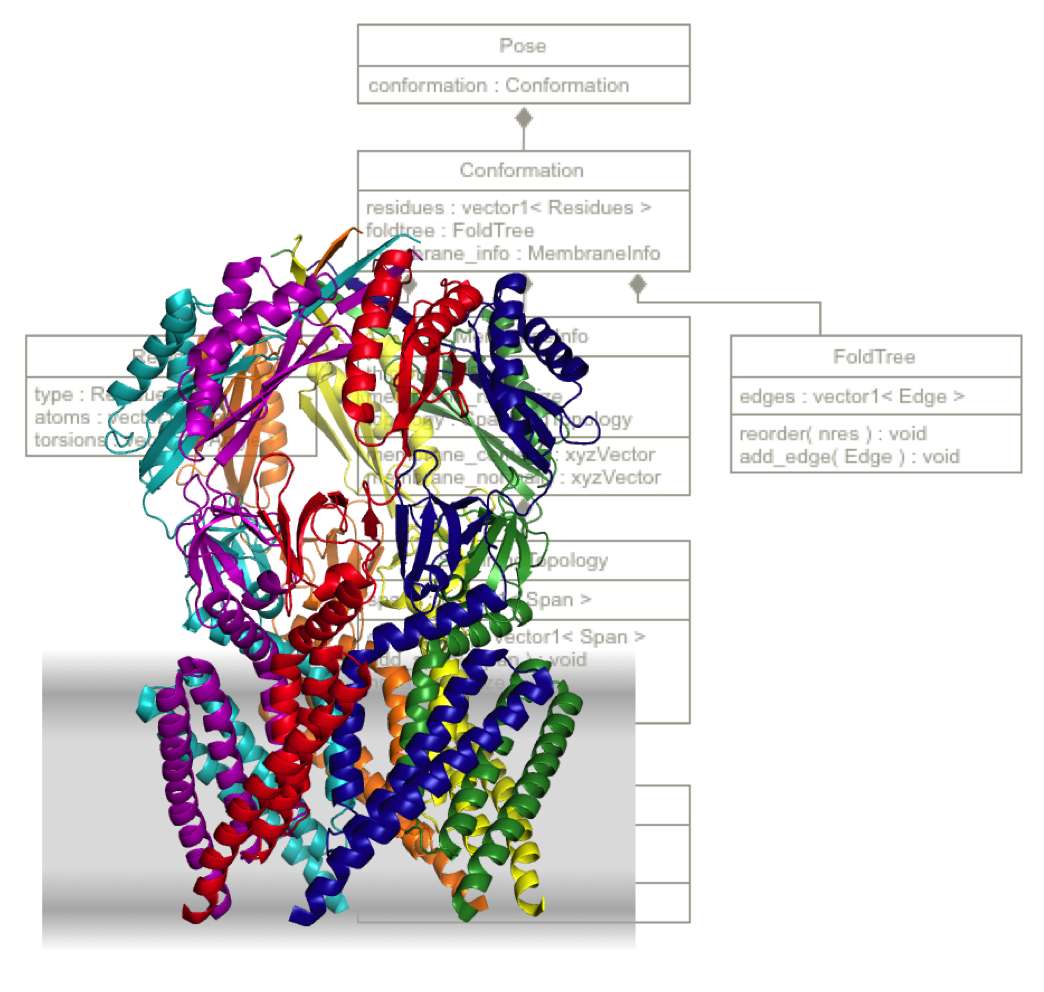
The major aspects of this implementation are the following:
MembraneInfo: MembraneInfo is a central hub for storing membrane-related data, including the membrane positon & orientation (center & normal), thickness, trans-membrane spans (SpanningTopology) and Liophilicity. Many of the sampling moves, score functions and protocols access MembraneInfo such that protocols can easily account for the membrane environment.
Membrane Representation: In contrast to the original RosettaMembrane, the membrane is represented in a special residue type (MEM) added to the pose. It stores the center point of the membrane, the membrane normal vector, and the thickness. The thickness value stored in MEM is half of the "true" membrane thickness, since the membrane is currently regarded as symmetric around z = 0 being at the center of the membrane. In the membrane coordinate system (right handed) the membrane is currently regarded as a flat membrane in the xy-plane with the membrane normal showing in the positive z direction.
Fixed and Moveable Membrane: In the RosettaMP framework, the membrane can be modeled as fixed or moveable during a simulation. While a flexible membrane is computationally cheaper and recommended for most protocols, some protocols require a fixed membrane for multiple poses to move in (such as MPDocking).
Energy Function: During the re-implementation of the Membrane Framework we kept the original low-resolution and high-resolution ScoreFunctions of RosettaMembrane.
Movers: Several Movers are available with more being implemented as needed. The most important one is the AddMembraneMover that creates a membrane pose from a regular pose - it requires a spanfile to be read in. When using Movers, please pay attention whether they are suited for your particular protocol, as many movers are specific to either (1) a fixed membrane and movable protein, or (2) a movable membrane and a fixed protein.
Visualization: The Membrane Framework is coupled to the PymolMover which allows real-time visualization of a Rosetta simulation with the membrane present and up-to-date.
Applications: Four main applications for membrane proteins (MPrelax, MPddG, MPdocking, symmetric MPdocking) are currently available with more to come. Not all applications are available for all interfaces, please see Alford, Koehler Leman et al. for more info, especially Figure 2 in this paper.
Contact
- Rebecca Alford (rfalford12@gmail.com)
- Julia Koehler Leman (julia.koehler1982@gmail.com)
- Corresponding PI: Jeffrey J. Gray (jgray@jhu.edu)
References
Main RosettaMP Reference
- Alford RF, Koehler Leman J, Weitzner BD, Duran AM, Tilley DC, Elazar A, et al. (2015) An Integrated Framework Advancing Membrane Protein Modeling and Design. PLoS Comput Biol 11(9): e1004398. (*equal contribution authors)
Rosetta All-atom membrane energy function
- Alford RF, Fleming PJ, Fleming KG, Gray JJ (2019) "Protein structure prediction and design in a biologically-realistic implicit membrane" BioRxiv
New Applications that use RosettaMP
- Koehler Leman J, Mueller BK, Gray JJ (2018) "Expanding the toolkit for membrane protein modeling in Rosetta" Bioinformatics 33 (5), 754-756
References for RosettaMembrane (mostly deprecated)
- Yarov-Yarovoy V, Schonbrun J, Baker D. (2006 Mar 1) Multipass membrane protein structure prediction using Rosetta. Proteins. 62(4):1010-25.
- Barth P, Schonbrun J, Baker D. (2007 Oct 2) Toward high-resolution prediction and design of transmembrane helical protein structures. Proc Natl Acad Sci U S A. 104(40):15682-7.
- Barth P, Wallner B, Baker D. (2009 Feb 3) Prediction of membrane protein structures with complex topologies using limited constraints. Proc Natl Acad Sci U S A. 106(5):1409-14.